The gut microbiota and drug metabolism
Overview
By Pr. Emily P. Balskus
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, USA
Lay public section
Find here your dedicated section
About this article
Author

Created
24 August 2021
Updated
22 July 2024
The gut microbiota transforms the chemical structures of ingested compounds, including orally-administered small molecule drugs. This metabolism, which can vary substantially between patients, impacts drug efficacy in both positive and negative ways, and can also influence toxicity. Over the last 10 years, there has been a growing appreciation of the potential contribution of gut microbiota drug metabolism to inter-individual variability in patient drug response. Here, we review this topic, with a focus on recent advances and their potential future impact on patient care and drug discovery.
The trillions of microorganisms that inhabit the human gut possess a greatly expanded set of genes compared to the host genome. Many of these genes encode protein-based catalysts, or enzymes, that enable gut microbes to perform a wide range of chemical reactions, expanding the chemistry associated with the human body. A hallmark of gut microbial metabolism is its variability; just as the composition of the microbiota differs between individuals, so too can the metabolic capabilities of this community. As we continue to identify associations between the gut microbiota and health and disease outcomes, it is becoming increasingly important to characterize microbial metabolic transformations at a molecular level.
One prominent activity associated with the gut microbiota is the ability to chemically modify the structures of small molecule drugs. [1] Orally administered drugs encounter gut microbes either prior to absorption in the small intestine or in the large intestine if they are poorly orally bioavailable. Orally administered or injected drugs, or drug metabolites, also reach the microbiota if they undergo biliary excretion into the intestine. Because a drug’s pharmacological activity directly arises from its chemical structure, microbial metabolism can have a large effect on drug action.
Effects of gut microbial drug metabolism
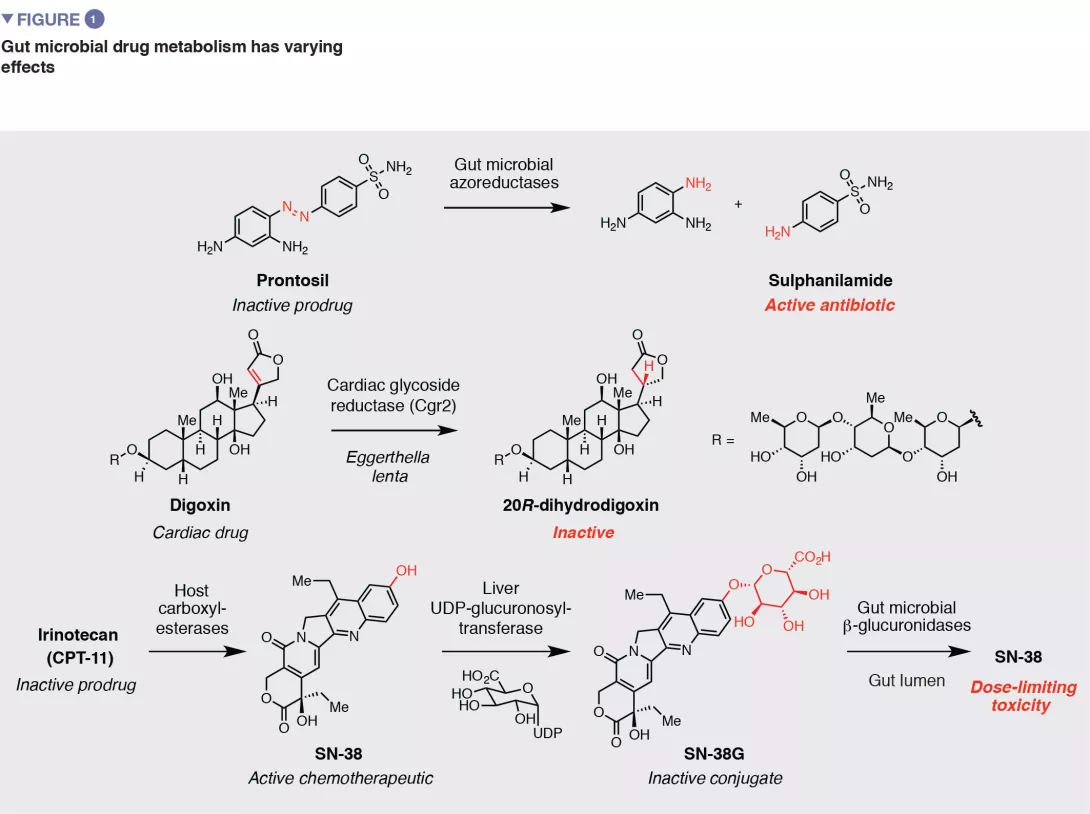
Gut microbial metabolism has various downstream consequences for drug action and efficacy (Figure 1). As the early examples of azo drugs illustrate, microbial metabolism of ‘prodrugs’ (inactive precursors) may be required to generate the active pharmacological agent. This knowledge has inspired the rational design of additional strategies for targeted drug release in the large intestine that rely on microbial metabolic activities Metabolism by the gut microbiota can also have negative effects on drug activity by disrupting interactions with intended host targets. One example is the natural product- based cardiac medication digoxin. In 5-10% of patients, the gut microbiota reduces the a, b-unsaturated lactone ring of digoxin to give dihydrodigoxin. This subtle modification, which is performed by the gut bacterium Eggerthella lenta, greatly reduces the binding affinity for digoxin’s target Na+/K+ ATPase, resulting in a loss of efficacy. [2] Another prominent example is the front-line Parkinson’s disease treatment L-dopa. Metabolism of L-dopa to dopamine by host enzymes in the brain is critical for alleviation of symptoms. Gut microbial metabolism of L-dopa also produces dopamine. [3,4] Because dopamine generated in the periphery cannot cross the blood brain barrier, this activity may reduce the amount of L-dopa that reaches the brain.
Finally, in addition to reducing activity, the chemical modifications installed by gut microbes can produce unwanted toxicity. For example, gut microbial metabolism was implicated in the lethality of co-administering the antiviral medication sorivudine with fluoropyrimidine chemotherapeutics. This outcome was traced to gut microbial metabolism of sorivudine to bromovinyluracil. This metabolite inhibits a key host enzyme involved in detoxifying 5-fluorouracil, increasing its concentration to lethal levels.
Studies of gut microbial drug metabolism began over 80 years ago with the discovery that the early antibiotic Prontosil, an azo compound that is inactive toward bacterial isolates but displays efficacy in vivo, underwent reduction by the gut microbiota to give the active agent sulfanilamide. Additional examples of gut microbial drug metabolism were uncovered throughout the intervening years, often prompted by observations of varying efficacy or toxicity in patients. Importantly, despite this history, such activities still are not typically considered in drug development or administration.
Image
An important characteristic of gut microbial drug metabolism is its variability across patients. This phenomenon has its origins in the variability of the gut microbiota. Though some metabolic activities are found in many organisms, others are carried out by a small, low abundant subset of the gut community. Metabolism can vary between individual strains of the same species, as even closely related bacteria can have large differences in their genomes. It is therefore perhaps unsurprising that community composition is often a poor predictor of metabolism, and metabolism of individual drugs can be extensive in some individuals and absent in others. This variation likely has important but incompletely understood consequences for patients taking a range of small molecule drugs.
Understanding drug metabolism at a molecular level
In order to fully understand gut microbial drug metabolism, it is necessary to link individual activities with microbes, genes, and enzymes. Identifying specific drug-metabolizing microbes is typically needed to enable downstream mechanistic studies. This may be accomplished through screening available gut microbial isolates or isolating metabolizing organisms directly from complex gut microbiota samples. An important next step is connecting transformations of interest to genes and enzymes. This is crucial for studying metabolism in complex gut communities, as the genes encoding metabolic enzymes allow detection and prediction of individual activities in microbial genomes and microbiome sequencing data. Linking drug metabolism to microbial genes can be accomplished in multiple ways, including rationally searching genomes for enzymes with the requisite catalytic capabilities, using RNA-Seq to identify genes that are specifically upregulated in response to a drug, and using comparative genomics to associate genes with metabolic capabilities.
Identifying new metabolic activities
Until 2019, approximately 60 examples of gut microbial drug metabolism were reported. Two recent studies leveraged approaches from high-throughput screening and experimentation to perform large scale surveys of gut microbial drug metabolism, greatly expanding the scope of known transformations. Goodman and co-workers screened 76 human gut bacterial isolates for their ability to metabolize 271 small molecule drugs and found that two thirds of the drugs were depleted by at least one organism. [8] The Donia group performed an analogous screen of 575 drugs using a patient gut microbiome sample ex vivo and uncovered 45 new transformations. [6] These efforts suggest the scope of drugs subject to metabolism may be larger than previously known; however, the vast majority of these newly reported activities have not yet been confirmed in vivo, so their relevance for patients is unknown.
The chemistry of gut microbial drug metabolism, which tends to be reductive and hydrolytic, is often unique from that of host transformations, which involve oxidation of drugs and conjugation with more polar metabolites to facilitate excretion. Microbial metabolism often has opposing effects on drug availability, prolonging circulation in the body. However, microbial drug transformations do not have to be distinct to impact drug action; recent studies of the anti-viral drug brivudine suggest such activities can affect drug pharmacokinetics even when they are identical to host metabolism. [5]
Manipulating gut microbial drug metabolism
Once the gut microbiota has been found to transform a small molecule drug, a logical next step is to ask how this activity may be controlled, both to assess the consequences of metabolism for drug action and to improve patient therapy should metabolism prove detrimental. Various methods have been employed to achieve this goal. Using gnotobiotic animal models (germfree animals colonized in a controlled manner with a defined microbiota), one can compare communities containing either drug metabolizing gut strains or deletion mutants missing specific activities. The utility of this approach was nicely illustrated by the Goodman lab’s studies of brivudine. [5]
However, genetic manipulation is challenging in native, complex microbial communities, prompting evaluation of alternative approaches. One potential strategy is to leverage knowledge of gut bacterial physiology to guide manipulation of the gut environment via dietary interventions. For example, digoxin Turnbaugh and co-workers noted that the presence of L-arginine downregulates drug metabolism by E. lenta. [2] They then showed that administering protein-rich diets to gnotobiotic mice colonized with E. lenta reduced drug inactivation in vivo.
Another exciting strategy is to identify small molecules that inhibit the activity of gut microbial drug metabolizing enzymes, as pioneered by the Redinbo lab in their studies of irinotecan metabolism. Irinotecan is a prodrug that is metabolized by host cells to the active topoisomerase inhibitor SN- 38. SN-38 is metabolized by the host via glucuronidation, which produces an inactive conjugate (SN-38G). This metabolite is excreted into the intestine, where the glucuronide is removed by gut bacterial b-glucoronidase (GUS) enzymes. This reactivation causes dose-limiting gastrointestinal tract toxicity. The Redinbo group used highthroughput screening to identify selective inhibitors of gut bacterial GUS enzymes, and found they prevented the severe side effects caused by irinotecan in a mouse model. [9] Subsequent work revealed that these compounds increase the efficacy of irinotecan by limiting its toxicity. [10] Together, this work has provided exciting proof-of-concept for therapeutically targeting gut bacterial metabolism and has prompted additional inhibitor discovery efforts.
An estimated 70% of gut microbial diversity is uncultivated, making it challenging to characterize their activities. Donia and co-workers used functional metagenomics, which introduces DNA isolated directly from a complex microbiota into a heterologous host, to identify a hydrocortisone-metabolizing gut bacterial enzyme. [6] Cholesterol metabolizing enzymes were also recently discovered in uncultured gut bacteria by correlating the presence of microbial genes in microbiomes with metabolomics data. [7] Both strategies may be useful for investigating drug metabolism by uncultured organisms.
Future frontiers
The successful development of GUS inhibitors as therapeutic candidates highlights one way in which gaining a molecular understanding of gut microbial drug metabolism could benefit patients. Another area that could be transformed by this knowledge is precision medicine. With an understanding of how specific therapeutics are metabolized by gut microbes, physicians could one day use microbiome sequencing data or microbiota-based diagnostic assays in deciding whether and how to prescribe particular medications.
Our growing appreciation of gut microbial drug metabolism may also influence the drug discovery process itself. Due to past associations with toxicity and side effects, many functional groups known to be transformed by gut bacteria are typically avoided by medicinal chemists. One could imagine uncovering new, unanticipated transformations early in drug development by screening individual gut microbes or complex patient communities for metabolism ex vivo, similarly to how drug candidates are typically tested for metabolism by host enzymes. Differences in gut microbiota composition and functions between animal models and humans should be taken into account in preclinical and clinical studies. Finally, it may be advisable to incorporate microbiome sample collection and analysis for drug metabolism into clinical trials. Correlating metabolism with differences in toxicity or efficacy might help in interpreting the results of such trials and defining target patient populations.
Sources