Gut mucosal and faeca l microbiota profiling combined to intestinal immune system in neonate s affected by intestinal ischemic injuries
Commented articles - Children's section
By Pr Emmanuel Mas
Gastroenterology and Nutrition Department, Children’s Hospital, Toulouse, France
Comments on the original article of Romani et al. (Frontiers in Cellular and Infection Microbiology 2020 [1])
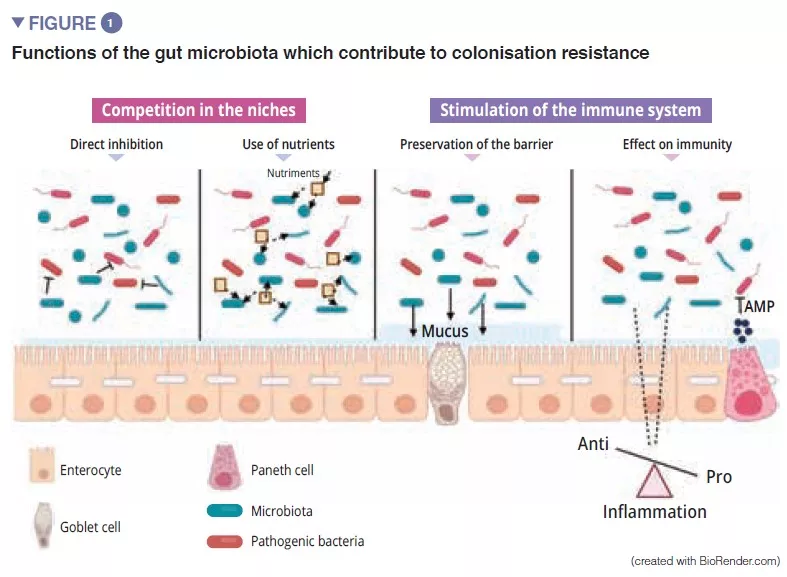
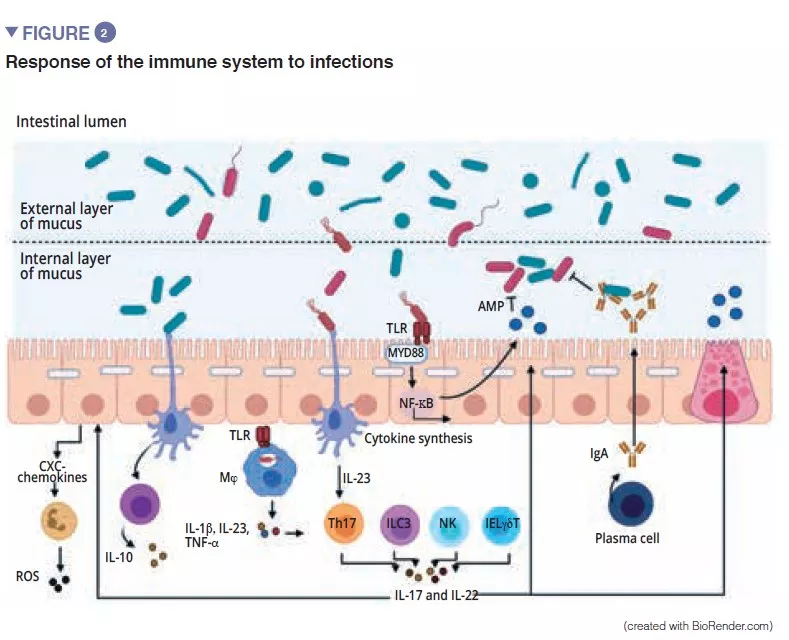
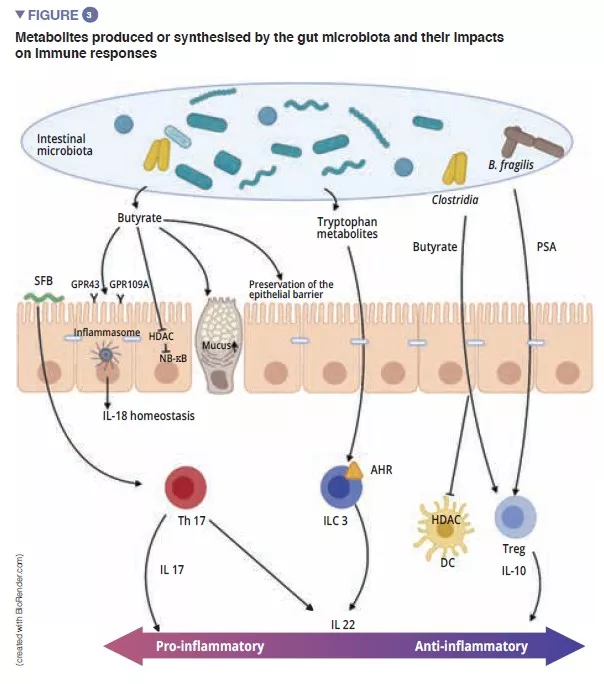
In the first weeks of life the microbiota plays a crucial role i n health by acting as a barrier against the invasion of pathogens and by maintaining intestinal immune homoeostasis. Changes in the ecology of the faecal microbiota (FM) have been reported in neonates with intestinal ischaemia. The aim of this study was to describe the FM, the mucosal microbiota (MM) and mucosal immunity in these patients.
Fourteen neonates underwent intestinal resection due to intestinal ischaemia. Two groups were identified on the basis of lesion severity: extensive (EII) and localised intestinal ischaemia (LII). This study showed the variations in FM and MM in the EII and LII groups.
What do we already know about this subject?
The intestinal microbiota of neonates is characterised by lower bacterial diversity and a higher proportion of pathogenic bacteria. Moreover, their intestinal immune system is immature. These two factors modify the intestinal epithelial barrier and enhance the production of pro-inflammatory mediators.
Necrotising enterocolitis (NEC) is an ischemic and inflammatory disorder of the gastro- intestinal tract which affects premature neonates. The physiopathology of NEC remains poorly understood but intestinal dysbiosis is present, and also an inflammatory process. The use of antibiotics and antacids promote dysbiosis and also increases the risk of onset of NEC.
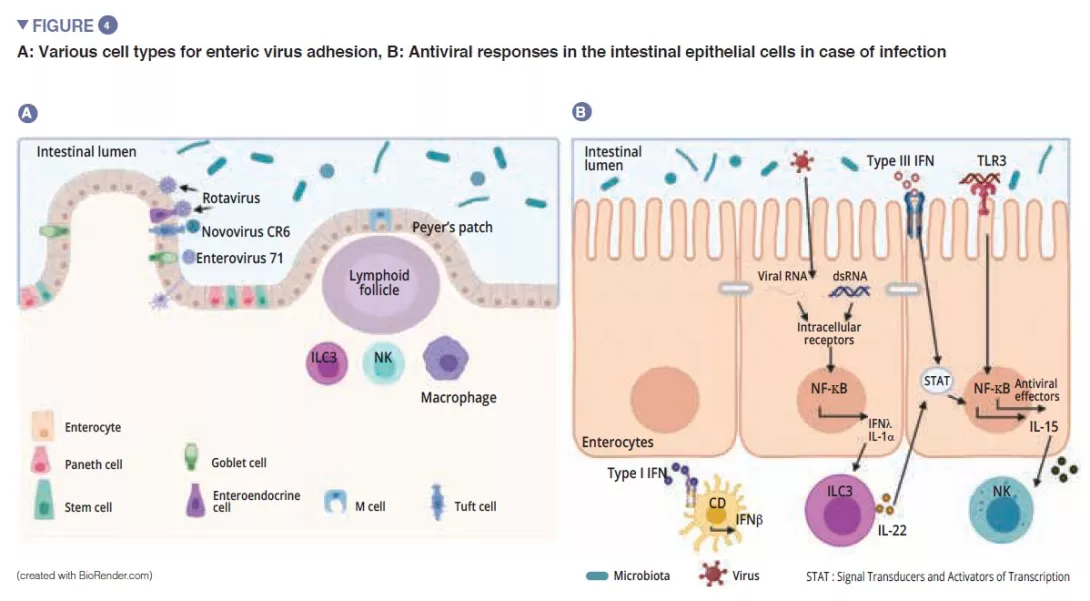
Neonates can also suffer from other ischaemic and inflammatory disorders, such as small bowel volvulus and localised gastro- intestinal perforations.
What are the main insights from this study?
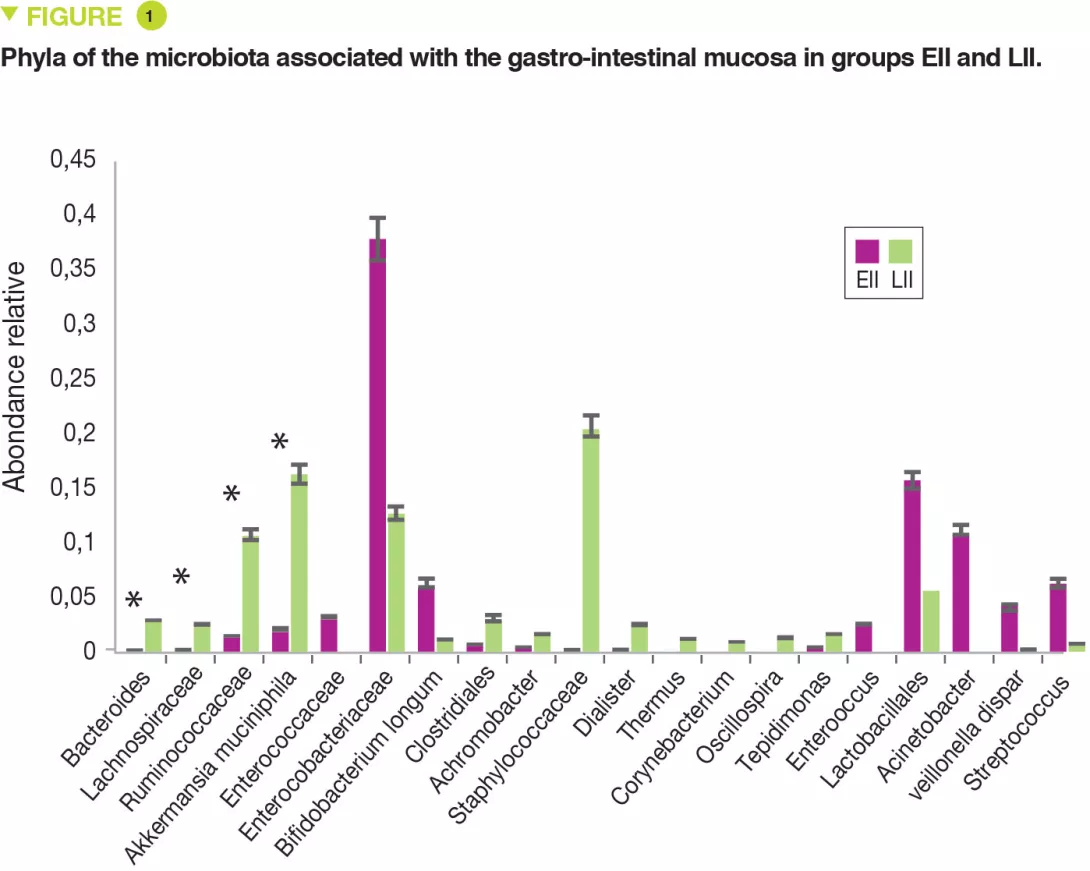
This single centre pilot study profiled the composition of the FM and MM and also the mononuclear cells of the lamina propria and the pro-inflammatory cytokines of two groups of neonates: full-term or premature. Seven infants had extensive intestinal ischaemia (EII) (5 NEC, 1 small bowel volvulus and 1 total colonic ischaemia) and 7 infants had localised intestinal ischaemia (LII) (4 isolated perforations and 3 cases of intestinal atresia). The FM of 9 full-term infants was used as control. The MM of neonates with EII, compared with those with LII, contained: more Proteobacteria (p = 0.049) and fewer Bacteroidetes (p = 0.007) and Verrucomicrobia (p = 0.01) (Figure 1); fewer Bacteroides, Lachnospiracee, Ruminococcaceae and Akkermansia muciniphila (p < 0.05).
The FM was less diverse (Shannon index) in EII than in LII (p = 0.01). The relative abundance for the MM was similar between EII and LII for Proteobacteria and Firmicutes (p < 0.05). Similarly, a bacterial distribution with more Enterobacteriaceae in EII and more Ruminococcaceae, Bacteroides, Lachnospiracee and Staphylococcaceae in cases of LII.
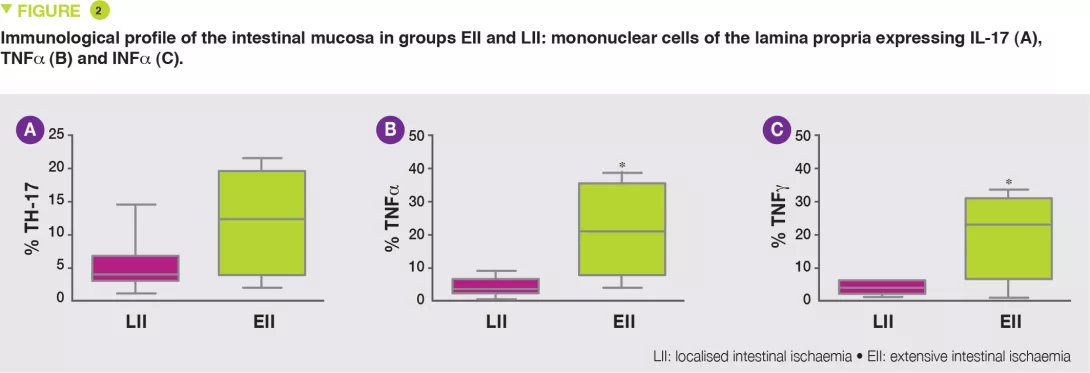
The EII group exhibited increased numbers of lymphocytes B, T and NK (Natural Killer), in particular T CD3+ lymphocytes, TH17 (Figure 2A) and reduced numbers of T lymphocyte regulators (Tregs), with increased numbers of cells expressing TNFa (Figure 2B) and INFg (Figure 2C).
What are the consequences in practice?
This pilot study confirms that lack of bacterial diversity and the predominance of Enterobacteriaceae are risk factors for NEC, as is a reduction in numbers of Akkermansia muciniphila. Correction of this dysbiosis could modify the TH17/Tregs imbalance and reduce the production of mediators of inflammation (TNFa and INFg).